Integrate and generate CITE-seq PBMC Datasets from scRNA-seq PBMC Datasets
The PBMC10k and PBMC5k CITE datasets was collected from Github. Ignore ADT measurements in the PBMC5k dataset and use only scRNA-seq data.
[1]:
import pandas as pd
import anndata
import numpy as np
import scanpy as sc
import scproca
from scproca.model import scProca
INFO Last run with scProca version: 0.1
Set global parameters
[2]:
scproca.settings.seed = 0
scproca.settings.batch_size = 512
scproca.settings.device = 0
INFO Setting seed 0
INFO Setting batch size 512
INFO Using cuda 0 device
[3]:
import seaborn as sns
import torch
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
%config InlineBackend.print_figure_kwargs={'facecolor' : "w"}
%config InlineBackend.figure_format='retina'
Load dataset from local source
[4]:
dataset_PBMC10k = anndata.read_h5ad("../../data/PBMC/pbmc_10k_protein_v3.h5ad")
dataset_PBMC10k.obs["batch"] = "PBMC10k"
dataset_PBMC5k = anndata.read_h5ad("../../data/PBMC/pbmc_5k_protein_v3.h5ad")
dataset_PBMC5k.obs["batch"] = "PBMC5k"
Standard data preprocessing
[5]:
common_genes = dataset_PBMC10k.var_names.intersection(dataset_PBMC5k.var_names)
dataset_PBMC10k = dataset_PBMC10k[:, common_genes]
dataset_PBMC5k = dataset_PBMC5k[:, common_genes]
dataset_PBMC10k.obsm["protein_expression"] = pd.DataFrame(
dataset_PBMC10k.obsm["protein_expression"],
columns=dataset_PBMC10k.uns["protein_names"],
index=dataset_PBMC10k.obs_names,
)
dataset_PBMC5k.obsm["protein_expression"] = pd.DataFrame(
dataset_PBMC5k.obsm["protein_expression"],
columns=dataset_PBMC5k.uns["protein_names"],
index=dataset_PBMC5k.obs_names,
)
del dataset_PBMC10k.uns["protein_names"]
del dataset_PBMC5k.uns["protein_names"]
adata = anndata.concat([dataset_PBMC10k, dataset_PBMC5k], join="inner", index_unique='-')
adata.obs_names_make_unique()
adata.obsm["protein_expression"] = adata.obsm["protein_expression"].fillna(0)
/tmp/ipykernel_35382/1631958525.py:4: ImplicitModificationWarning: Setting element `.obsm['protein_expression']` of view, initializing view as actual.
dataset_PBMC10k.obsm["protein_expression"] = pd.DataFrame(
/tmp/ipykernel_35382/1631958525.py:9: ImplicitModificationWarning: Setting element `.obsm['protein_expression']` of view, initializing view as actual.
dataset_PBMC5k.obsm["protein_expression"] = pd.DataFrame(
Adapt data for scProca
Ignore ADT measurements in the PBMC5k dataset and use only scRNA-seq data.
[6]:
adata = adata[np.random.permutation(adata.n_obs)].copy()
batch = adata.obs["batch"].values.ravel()
held_out_proteins_PBMC5k = adata.obsm["protein_expression"][batch == "PBMC5k"].copy()
adata.obsm["protein_expression"].loc[batch == "PBMC5k"] = np.zeros(
adata.obsm["protein_expression"][batch == "PBMC5k"].shape
)
adata.obs["batch_general"] = adata.obs["batch"].map({
"PBMC10k" : "CITE-seq (PBMC10k)",
"PBMC5k" : "scRNA-seq (PBMC5k)"
})
valid_adt = np.array([True] * len(adata))
valid_adt[batch == "PBMC5k"] = False
adata.obs["valid_adt"] = valid_adt
sc.pp.highly_variable_genes(
adata,
n_top_genes=4000,
flavor="seurat_v3",
batch_key="batch",
subset=True
)
[7]:
adata
[7]:
AnnData object with n_obs × n_vars = 10849 × 4000
obs: 'n_genes', 'percent_mito', 'n_counts', 'batch', 'batch_general', 'valid_adt'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'hvg'
obsm: 'protein_expression'
Initialize scProca model
[8]:
scproca_model = scProca(
adata=adata,
key_adt="protein_expression",
key_batch="batch",
key_valid_adt="valid_adt"
)
INFO Computing empirical prior initialization for protein background.
Train scProca model
[9]:
scproca_model.train()
scproca_model.curve_loss(key_loss="loss_elbo")
scproca_model.curve_loss(key_loss="loss_discriminator")
/data/data2/xionglab/anaconda3/envs/scMuMA/lib/python3.11/site-packages/torch/optim/lr_scheduler.py:28: UserWarning: The verbose parameter is deprecated. Please use get_last_lr() to access the learning rate.
warnings.warn("The verbose parameter is deprecated. Please use get_last_lr() "
Training Progress: 65%|███████████▋ | 261/400 [04:26<02:21, 1.02s/it, loss_discriminator=1.94, loss_elbo=2.41e+3]
Early stopping triggered.
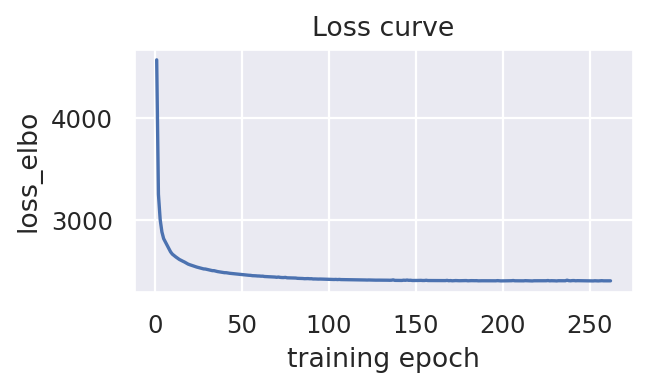
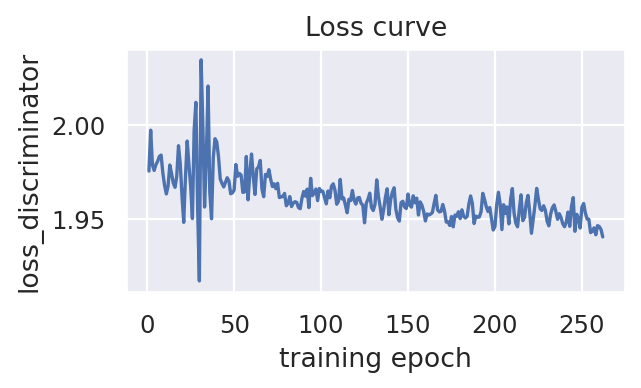
Infer the integrated latent representation, RNA-specific embedding, and ADT-specific embedding for each cell
[10]:
adata.obsm["latent"], adata.obsm["embedding_rna"], adata.obsm["embedding_adt"] = scproca_model.get_latent_representation()
Inference Progress: 100%|█████████████████████████████████████████████████████████████| 100/100 [00:13<00:00, 7.25it/s]
[11]:
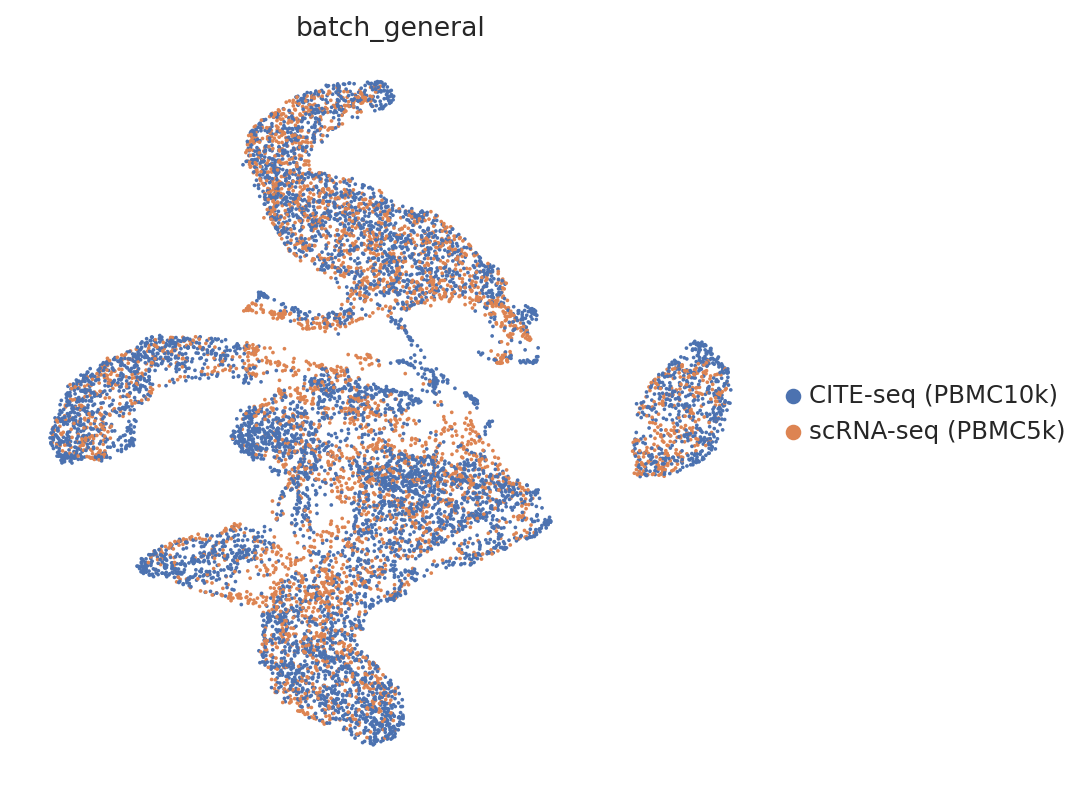
sc.pp.neighbors(adata, use_rep="embedding_rna")
sc.tl.umap(adata)
sc.pl.umap(
adata,
color="batch_general",
frameon=False
)

[12]:
sc.pp.neighbors(adata, use_rep="embedding_adt")
sc.tl.umap(adata)
sc.pl.umap(
adata,
color="batch_general",
frameon=False
)
[13]:
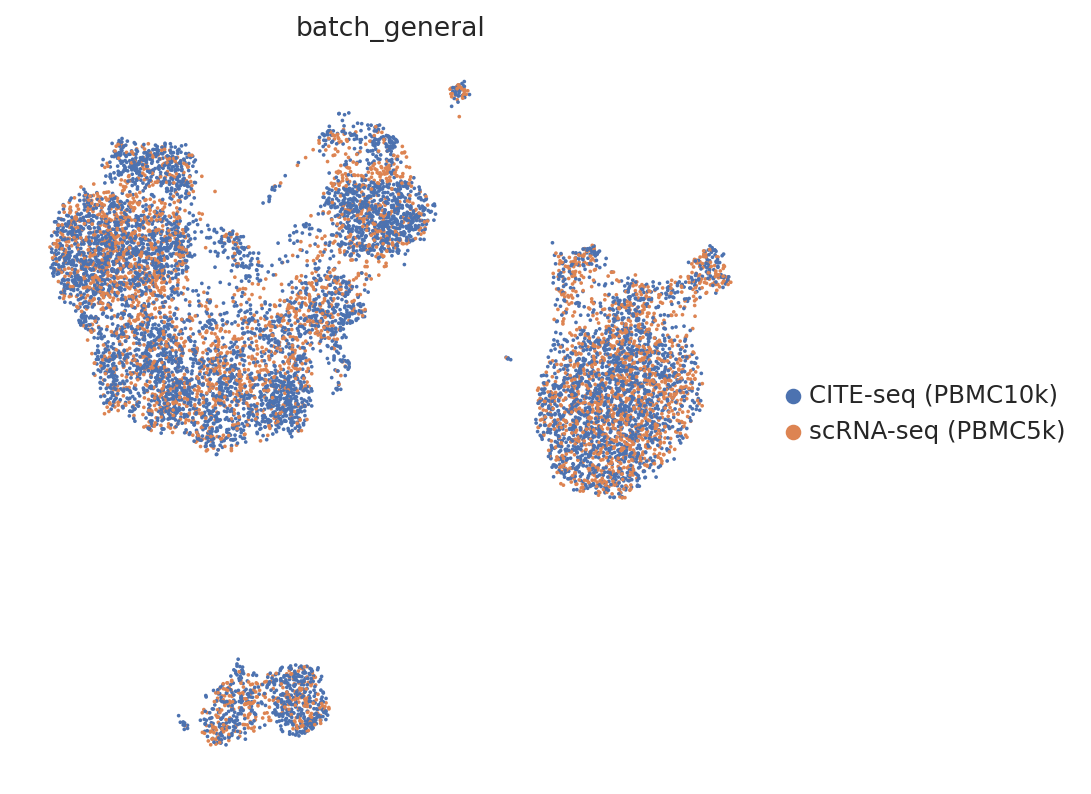
sc.pp.neighbors(adata, use_rep="latent")
sc.tl.umap(adata)
sc.pl.umap(
adata,
color="batch_general",
frameon=False
)
Generate ADT measurements for each cell
[14]:
protein_generation = scproca_model.generation(anchor_batch="PBMC10k")
protein_observed = adata.obsm["protein_expression"].values.copy()
protein_observed[batch == "PBMC5k"] = held_out_proteins_PBMC5k.values
# cleaner protein names
parsed_protein_names = [
p.split("_")[0] for p in adata.obsm["protein_expression"].columns
]
for i, p in enumerate(parsed_protein_names):
adata.obs[f"{p} imputed"] = protein_generation[:, i]
adata.obs[f"{p} observed"] = protein_observed[:, i]
Inference Progress: 100%|█████████████████████████████████████████████████████████████| 100/100 [00:15<00:00, 6.30it/s]
[15]:
viz_keys = []
for p in parsed_protein_names:
viz_keys.append(p + " imputed")
viz_keys.append(p + " observed")
sc.pl.umap(
adata[batch == "PBMC5k"],
color=viz_keys,
ncols=2,
vmax="p99",
frameon=False,
add_outline=True,
wspace=0.1,
)
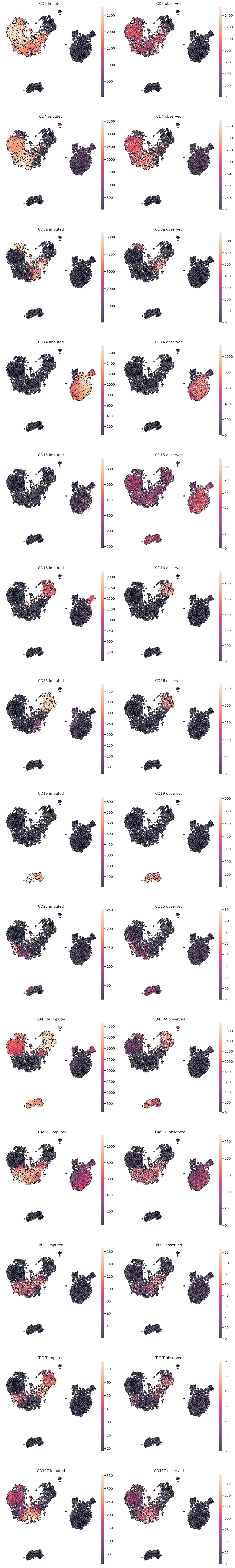
[15]: